A wrapper around the vegan package to generate ggplot2 ordination plots suited for analysis and comparison of microbial communities. Simply choose an ordination type and a plot is returned.
amp_ordinate(
data,
filter_species = 0.1,
type = "PCA",
distmeasure = "bray",
transform = "hellinger",
constrain = NULL,
x_axis = 1,
y_axis = 2,
print_caption = FALSE,
sample_color_by = NULL,
sample_color_order = NULL,
sample_shape_by = NULL,
sample_colorframe = FALSE,
sample_colorframe_label = NULL,
sample_colorframe_label_size = 3,
sample_label_by = NULL,
sample_label_size = 4,
sample_label_segment_color = "black",
sample_point_size = 2,
sample_trajectory = NULL,
sample_trajectory_group = NULL,
sample_plotly = NULL,
species_plot = FALSE,
species_nlabels = 0,
species_label_taxonomy = "Genus",
species_label_size = 3,
species_label_color = "grey10",
species_rescale = FALSE,
species_point_size = 2,
species_shape = 20,
species_plotly = FALSE,
envfit_factor = NULL,
envfit_numeric = NULL,
envfit_signif_level = 0.005,
envfit_textsize = 3,
envfit_textcolor = "darkred",
envfit_numeric_arrows_scale = 1,
envfit_arrowcolor = "darkred",
envfit_show = TRUE,
repel_labels = TRUE,
opacity = 0.8,
tax_empty = "best",
detailed_output = FALSE,
num_threads = 1L,
...
)Arguments
- data
(required) Data list as loaded with
amp_load.- filter_species
Remove low abundant OTU's across all samples below this threshold in percent. Setting this to 0 may drastically increase computation time. (default:
0.1)- type
(required) Type of ordination method. One of:
"PCA": (default) Principal Components Analysis"RDA": Redundancy Analysis (considered the constrained version of PCA)"CA": Correspondence Analysis"CCA": Canonical Correspondence Analysis (considered the constrained version of CA)"DCA": Detrended Correspondence Analysis"NMDS": non-metric Multidimensional Scaling"PCOA"or"MMDS": metric Multidimensional Scaling a.k.a Principal Coordinates Analysis (not to be confused with PCA)
Note that PCoA is not performed by the vegan package, but the
pcoafunction from the APE package.- distmeasure
(required for nMDS and PCoA) Distance measure used for the distance-based ordination methods (nMDS and PCoA). Choose one of the following:
"wunifrac"(PCoA only): Weighted UniFrac distances. Requires a rooted phylogenetic tree."unifrac"(PCoA only): Unweighted UniFrac distances. Requires a phylogenetic tree."jsd"(PCoA only): Jensen-Shannon Divergence, based on http://enterotype.embl.de/enterotypes.html.Any of the distance measures supported by
vegdist:"manhattan","euclidean","canberra","bray","kulczynski","gower","morisita","horn","mountford","jaccard","raup","binomial","chao","altGower","cao","mahalanobis","clark","chisq","chord","hellinger","aitchison","robust.aitchison".
You can also write your own math formula, see details in
vegdist. Default isbray.- transform
(recommended) Transforms the abundances before ordination, choose one of the following:
"total","max","freq","normalize","range","standardize","pa"(presence/absense),"chi.square","hellinger","log", or"sqrt", see details indecostand. Using the hellinger transformation is a good choice when performing PCA/RDA as it will produce a more ecologically meaningful result (read about the double-zero problem in Numerical Ecology). When the Hellinger transformation is used with CA/CCA it will help reducing the impact of low abundant species. When performing nMDS or PCoA (aka mMDS) it is not recommended to also use data transformation as this will obscure the chosen distance measure. (default:"hellinger")- constrain
(required for RDA and CCA) Variable(s) in the metadata for constrained analyses (RDA and CCA). Multiple variables can be provided by a vector, fx
c("Year", "Temperature"), but keep in mind that the more variables selected the more the result will be similar to unconstrained analysis.- x_axis
(integer) Which axis from the ordination results to plot as the first axis. Have a look at the
$screeplotwithdetailed_output = TRUEto validate axes. With nMDS the number of dimensions (kargument to themetaMDSfunction) is set to that of the highest number of eitherx_axisory_axis. (default:1)- y_axis
(integer) Which axis from the ordination results to plot as the second axis. Have a look at the
$screeplotwithdetailed_output = TRUEto validate axes. With nMDS the number of dimensions (kargument to themetaMDSfunction) is set to that of the highest number of eitherx_axisory_axis. (default:2)- print_caption
Auto-generate a figure caption based on the arguments used. The caption includes a description of how the result has been generated as well as references for the methods used.
- sample_color_by
Color sample points by a variable in the metadata.
- sample_color_order
Order the colors in
sample_color_byby the order in a vector.- sample_shape_by
Shape sample points by a variable in the metadata.
- sample_colorframe
Frame the sample points with a polygon by a variable in the metadata split by the variable defined by
sample_color_by, or simplyTRUEto frame the points colored bysample_color_by. (default:FALSE)- sample_colorframe_label
Label by a variable in the metadata.
- sample_colorframe_label_size
Size of the color frame labels. (default:
3)- sample_label_by
Label sample points by a variable in the metadata.
- sample_label_size
Sample labels text size. (default:
4)- sample_label_segment_color
Sample labels repel-segment color. (default:
"black")- sample_point_size
Size of the sample points. (default:
2)- sample_trajectory
Make a trajectory between sample points by a variable in the metadata.
- sample_trajectory_group
Make a trajectory between sample points by the
sample_trajectoryargument, but within individual groups.- sample_plotly
Enable interactive sample points so that they can be hovered to show additional information from the metadata. Provide a vector of the metadata variables to show, or
"all"to display all. Click or double click the elements in the legend to hide/show parts of the data. To hide the legend useplotly::layout(amp_ordinate(...), showlegend = FALSE), see more options at https://plot.ly/r/.- species_plot
(logical) Plot species points or not. (default:
FALSE)- species_nlabels
Number of the most extreme species labels to plot (ordered by the sum of the numerical values of the x,y coordinates. Only makes sense with PCA/RDA).
- species_label_taxonomy
Taxonomic level by which to label the species points. (default:
"Genus")- species_label_size
Size of the species text labels. (default:
3)- species_label_color
Color of the species text labels. (default:
"grey10")- species_rescale
(logical) Rescale species points or not. Basically they will be multiplied by 0.8, for visual convenience only. (default:
FALSE)- species_point_size
Size of the species points. (default:
2)- species_shape
The shape of the species points, fx
1for hollow circles or20for dots. (default:20)- species_plotly
(logical) Enable interactive species points so that they can be hovered to show complete taxonomic information. (default:
FALSE)- envfit_factor
A vector of categorical environmental variables from the metadata to fit onto the ordination plot. See details in
envfit.- envfit_numeric
A vector of numerical environmental variables from the metadata to fit arrows onto the ordination plot. The lengths of the arrows are scaled by significance. See details in
envfit.- envfit_signif_level
The significance threshold for displaying the results of
envfit_factororenvfit_numeric. (default:0.005)- envfit_textsize
Size of the envfit text on the plot. (default:
3)- envfit_textcolor
Color of the envfit text on the plot. (default:
"darkred")- envfit_numeric_arrows_scale
Scale the size of the numeric arrows. (default:
1)- envfit_arrowcolor
Color of the envfit arrows on the plot. (default:
"darkred")- envfit_show
(logical) Show the results on the plot or not. (default:
TRUE)- repel_labels
(logical) Repel all labels to prevent cluttering of the plot. (default:
TRUE)- opacity
Opacity of all plotted points and sample_colorframe.
0: invisible,1: opaque. (default:0.8)- tax_empty
How to show OTUs without taxonomic information. One of the following:
"remove": Remove OTUs without taxonomic information."best": (default) Use the best classification possible."OTU": Display the OTU name.
- detailed_output
(logical) Return additional details or not (model, scores, figure caption, inputmatrix, screeplot etc). If
TRUE, it is recommended to save to an object and then access the additional data byView(object$data). (default:FALSE)- num_threads
The number of threads to use whereever in the code parallelisation is possible. Any parallel computation is being performed by using
foreach. (default:1)- ...
Pass additional arguments to the vegan ordination functions, fx the
rda,cca,metaMDSfunctions, see the documentation.
Value
A ggplot2 object. If detailed_output = TRUE a list with a ggplot2 object and additional data.
Details
The amp_ordinate function is primarily based on two packages; vegan-package, which performs the actual ordination, and the ggplot2-package to generate the plot. The function generates an ordination plot by the following process:
Various input argument checks and error messages
OTU-table filtering, where low abundant OTU's across all samples are removed (if not
filter_species = 0is set)Data transformation (if not
transform = "none"is set)Calculate distance matrix based on the chosen
distmeasureif the chosen ordination method is PCoA/nMDS/DCAPerform the actual ordination and calculate the axis scores for both samples and species/OTU's
Visualise the result with ggplot2 or plotly in various ways defined by the user
When the chosen ordination method is an eigenanalysis-based method then the relative contribution (eigenvalue) of each axis to the total inertia in the data (sum of all eigenvalues, including those of the constrained space) is indicated in percent at the axis titles. When one of the constrained ordination methods (RDA and CCA) is used then a second value is furthermore shown which then indicates the relative contribution of the particular axis to the total constrained space only.
Using a custom distance matrix
If you wan't to calculate a distance matrix manually and use it for PCoA or nMDS in amp_ordinate, it can be done by setting filter_species = 0, transform = "none", distmeasure = "none", and then override the abundance table ($abund) in the ampvis2 object, like below. The matrix must be a symmetrical matrix containing coefficients for all pairs of samples in the data.
#Override the abundance table in the ampvis2 object with a custom distance matrix
ampvis2_object$abund <- custom_dist_matrix
#set filter_species = 0, transform = "none", and distmeasure = "none"
amp_ordinate(ampvis2_object,
type = "pcoa",
filter_species = 0,
transform = "none",
distmeasure = "none")References
GUide to STatistical Analysis in Microbial Ecology (GUSTA ME): https://mb3is.megx.net/gustame
Legendre, Pierre & Legendre, Louis (2012). Numerical Ecology. Elsevier Science. ISBN: 9780444538680
Legendre, P., & Gallagher, E. (2001). Ecologically meaningful transformations for ordination of species data. Oecologia, 129(2), 271-280. doi:10.1007/s004420100716
See also
Examples
# Load example data
data("AalborgWWTPs")
# PCA with data transformation, colored by WWTP
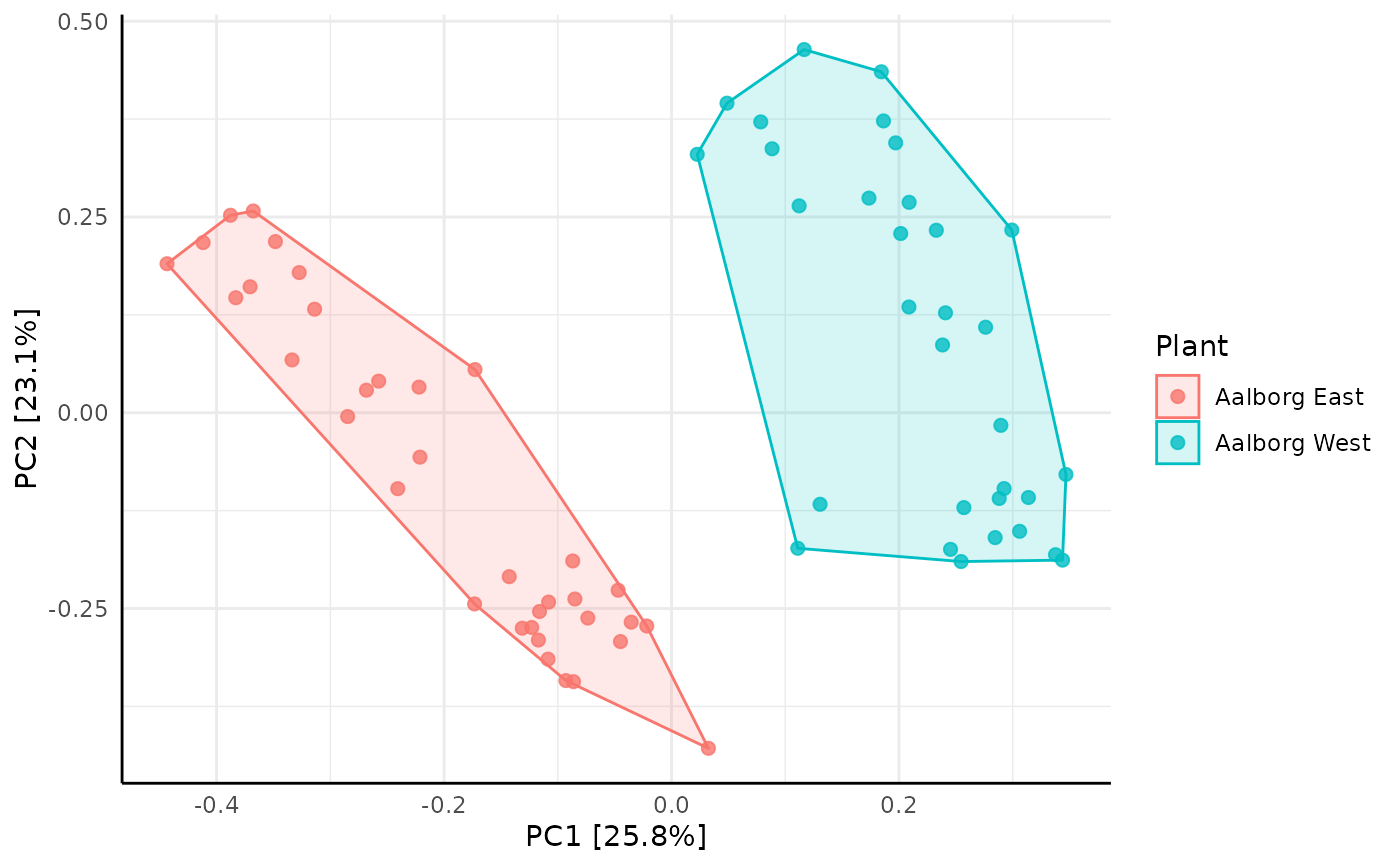
amp_ordinate(AalborgWWTPs,
type = "PCA",
transform = "hellinger",
sample_color_by = "Plant",
sample_colorframe = TRUE
)
#> 8628 OTUs not present in more than 0.1% relative abundance in any sample have been filtered
#> Before: 9430 OTUs
#> After: 802 OTUs
#> Warning: The `size` argument of `element_line()` is deprecated as of ggplot2 3.4.0.
#> ℹ Please use the `linewidth` argument instead.
#> ℹ The deprecated feature was likely used in the ampvis2 package.
#> Please report the issue at <https://github.com/kasperskytte/ampvis2/issues>.
 if (FALSE) { # \dontrun{
# Interactive CCA with data transformation constrained to seasonal period
amp_ordinate(AalborgWWTPs,
type = "CCA",
transform = "Hellinger",
constrain = "Period",
sample_color_by = "Period",
sample_colorframe = TRUE,
sample_colorframe_label = "Period",
sample_plotly = "all"
)
} # }
if (FALSE) { # \dontrun{
# Interactive CCA with data transformation constrained to seasonal period
amp_ordinate(AalborgWWTPs,
type = "CCA",
transform = "Hellinger",
constrain = "Period",
sample_color_by = "Period",
sample_colorframe = TRUE,
sample_colorframe_label = "Period",
sample_plotly = "all"
)
} # }